CASES AUTH/2898/11/16 AND AUTH/2901/11/16 DIRECTOR v ROCHE
Clinical trial disclosure (Kadcyla and Perjeta)
A study published online in Current Medical Research & Opinion (CMRO) on 25 November 2016 was entitled ‘Clinical trial transparency update: an assessment of the disclosure of results of company-sponsored trials associated with new medicines approved in Europe in 2013’. The study authors were B R Deane, a freelance consultant in pharmaceutical marketing and research and Dr J Sivarajah, Head of Medical Affairs, ABPI. Publication support for the study was funded by the ABPI.
The study surveyed various publicly available information sources for clinical trial registration and disclosure of results searched between 1 May and 31 July 2015. It covered 34 new medicines (except vaccines) from 24 companies that were approved by the European Medicines Agency (EMA) in 2013. It included all completed company-sponsored clinical trials conducted in patients and recorded on a clinical trial registry and/or included in a European Public Assessment Report (EPAR). The CMRO publication did not include the specific data for each product. This was available in the supplemental information via a website link. Neither the study nor the supplemental information identified specific clinical trials. The study did not assess the content of disclosure against any specific requirements.
The Director decided that the study was such that she had received information from which it appeared that Roche might have breached the Code and decided in accordance with Paragraph 5.1 of the Constitution and Procedure to take the matter up as a complaint.
The summary output for each medicine set out the sources for all trials found, irrespective of sponsor and an analysis of publication disclosure in the form of a table which gave details for the studies for Kadcyla (trastuzumab emtansine) and Perjeta (pertuzumab).
The detailed response from Roche is given below.
General detailed comments from the Panel are given below.
With regard to Kadcyla, the Panel noted the CMRO publication in that one evaluable trial had not been disclosed within the timeframe. The disclosure percentage at 12 months measured from the later of the first date of regulatory approval or trial completion date was 91%. The disclosure percentage at 31 July 2015 was 91%.
Kadcyla was first approved and commercially available in February 2013.
With regard to Perjeta, the Panel noted the CMRO publication in that one evaluable trial had not been disclosed within the timeframe. The disclosure percentage at 12 months was 95%. The disclosure percentage at 31 July 2015 was 100%.
Perjeta was first approved and commercially available in June 2012.
The Panel noted Roche’s submission that the alleged undisclosed trial in each case related to one Phase Ib/IIa study which included both Kadcyla and Perjeta. The trial was conducted in multiple sites by Roche global and included one UK trial site and thus fell within the scope of the ABPI Code with regard to disclosure as acknowledged by Roche.
The Panel considered that the Second 2012 Code and the Joint Position 2009 applied based on the first commercialisation of Kadcyla.
The trial completed on 24 October 2013 which was after the date of commercialisation for both Kadcyla and Perjeta. The Panel noted that on the information before it the trial results should have been posted by 24 October 2014. The Panel noted Roche’s submission that the trial at issue was registered on ClinicalTrials.gov on 6 July 2009 however due to an incorrect Phase I categorisation (rather than Phase I/II) within Roche, results were not posted to ClinicalTrials.gov. The trial had now been reclassified within Roche.
The Panel noted that data from the trial was published at the San Antonio Breast Cancer Symposium, in December 2012 (interim analysis) and December 2013, however the complete results had not been posted on a publicly accessible, internet based, clinical trials database within the required timeframe as acknowledged by Roche. The Panel thus ruled a breach of the Code. The delay in disclosure meant that high standards had not been maintained and a breach of the Code was ruled.
As the data had now been disclosed the Panel considered that there was no breach of Clause 2 and ruled accordingly.
A study published online in Current Medical Research & Opinion (CMRO) on 25 November 2016 was entitled ‘Clinical trial transparency update: an assessment of the disclosure of results of company sponsored trials associated with new medicines approved in Europe in 2013’. The study authors were B R Deane, a freelance consultant in pharmaceutical marketing and research and Dr J Sivarajah, Head of Medical Affairs, ABPI. Publication support for the study was funded by the ABPI.
The study referred to the two previously reported studies which covered medicines approved in Europe in 2009, 2010 and 2011 (Rawal and Deane 2014) and in 2012 (Rawal and Deane 2015). The 2016 study surveyed various publicly available information sources for clinical trial registration and disclosure of results searched between 1 May and 31 July 2015. It covered 34 new medicines (except vaccines) from 24 companies that were approved by the European Medicines Agency (EMA) in 2013. It included all completed company sponsored clinical trials conducted in patients and recorded on a clinical trial registry and/or included in a European Public Assessment Report (EPAR). The CMRO publication did not include the specific data for each product. This was available in the supplemental information via a website link. Neither the study nor the supplemental information identified specific clinical trials. The CMRO study did not assess the content of disclosure against any specific requirements.
The Director decided that the study was such that she had received information from which it appeared
Kadcyla
that Roche might have breached the Code and so she decided in accordance with Paragraph 5.1 of the Constitution and Procedure to take the matter up as a complaint.
COMPLAINT
The study assessed the proportion of trials for which results had been disclosed on a registry or in the scientific literature either within 12 months of the later of either first regulatory approval or trial completion, or by 31 July 2015 (end of survey). Of the completed trials associated with 34 new medicines licensed to 24 different companies in 2013, results of 90% (484/539) had been disclosed within 12 months and results of 93% (500/539) had been disclosed by 31 July 2015.
The supplemental information gave details of disclosure of clinical trial results for each product irrespective of sponsor. The data for Kadcyla (trastuzumab emtansine) were as follows:
|
Phase
|
Total
|
Unevaluable
|
Evaluable
|
Disclosed in 12-month timeframe
|
Disclosure Percentage
|
Complete before 31
July 2015
|
Disclosed at 31 July
2015
|
Disclosure percentage at 31 July
2015
|
|
Phase I & II
Phase III Phase IV
Other
|
11
2
0
0
|
2
0
0
0
|
9
2
0
0
|
8
2
0
0
|
89%
100%
|
9
2
0
0
|
8
2
0
0
|
89%
100%
|
|
Total
|
13
|
2
|
11
|
10
|
91%
|
11
|
10
|
91%
|
Footnote (company communication): Results of one phase I trial (originally put of scope of disclosure requirements) remained undisclosed. The results have been submitted for publication and will be posted on EudraCT.
Perjeta
The supplemental information gave details of disclosure of clinical trial results for each product irrespective of sponsor. The data for Perjeta (pertuzumab) were as follows:
|
Phase
|
Total
|
Unevaluable
|
Evaluable
|
Disclosed in 12-month timeframe
|
Disclosure Percentage
|
Complete before 31
July 2015
|
Disclosed at 31 July
2015
|
Disclosure percentage at 31 July
2015
|
|
Phase I & II
Phase III
Phase IV
Other
|
20
1
0
0
|
0
0
0
0
|
20
1
0
0
|
19
1
0
0
|
95%
100%
|
20
1
0
0
|
20
1
0
0
|
100%
100%
|
|
Total
|
21
|
0
|
21
|
20
|
95%
|
21
|
21
|
100%
|
The explanation of terms given in the documentation was as follows:
|
Total
|
Total number of company sponsored trials identified which were completed by 31 July 2015
|
|
Unevaluable
|
Trials with completion date within the last 12 months or key dates missing – excluded from the analysis
|
|
Evaluable
|
Trials with all criteria present including dates, and hence the base number of trials which could be evaluated for the assessment
|
|
Results disclosed in 12 month timeframe
|
Evaluable trials which were disclosed within the target 12 months measured from the later of either first regulatory approval date in Europe or the US, or trial completion date
|
|
Disclosure percentage
|
Proportion of evaluable trials which were disclosed within 12 months measured from the later of either first regulatory approval date in Europe or the US, or trial completion date
|
|
Completed before 31 July 2015
|
Number of evaluable trials completed before 31 July 2015
|
|
Disclosed at 31 July 2015
|
Number of evaluable trials with results disclosed by 31 July 2015
|
|
Disclosure percentage at 31 July 2015
|
Proportion of evaluable trials which were disclosed by 31 July 2015
|
When writing to Roche the Authority asked it to bear in mind the requirements of Clauses 2, 9.1 and 13.1 of the Code. The Authority noted that previous editions of the Code would be relevant and provided details.
RESPONSE
Roche submitted that it recognised the importance of accurate and timely disclosure and remained committed to broadening access to its clinical data.
Roche stated that it had a high degree of governance around data transparency for clinical trials. The company’s policy and commitment was to ensure publication of clinical trial data in peer-reviewed journals and on publicly available clinical trial registries of the US National Institutes of Health (NIH) and the European Medicines Agency (EMA). As detailed in the Global Policy on Sharing of Clinical Trials Data, Roche also granted requests for access to full clinical study reports, periodic safety reports and summary reports of clinical data across multiple trials, upon request. The company’s standard operating procedure (SOP) relating to Global Clinical trials disclosures and the Global Publication Policy described the process that underpinned its data sharing policy. The UK affiliate was within the scope of all of these documents.
Roche stated that Kadcyla as a single agent was indicated for the treatment of adults with human epidermal growth factor receptor (HER2)-positive, unresectable locally advanced or metastatic breast cancer who previously received trastuzumab and a taxane, separately or in combination. Patients should have either received prior therapy for locally advanced or metastatic disease; or developed disease recurrence during or within six months of completing adjuvant therapy. Kadcyla was first licensed and commercialised in the US on 22 February 2013 and subsequently approved in the EU on 15 November 2013.
Roche stated that Perjeta was indicated for use in combination with trastuzumab and docetaxel in adults with HER2-positive metastatic or locally recurrent unresectable breast cancer, who had not received previous anti-HER2 therapy or chemotherapy for their metastatic disease. It was also indicated for use in combination with trastuzumab and chemotherapy for the neoadjuvant treatment of adults with HER2-positive, locally advanced, inflammatory, or early stage breast cancer at high risk of recurrence. It was first licensed and commercialised in the US on 8 June 2012, with first approval in the EU on 4 March 2013.
Roche noted that one trial each for Kadcyla and Perjeta was not disclosed within the required 12 month timeframe nor disclosed by 31 July 2015 (the end of the CMRO study). Roche submitted that in both of these instances, the alleged undisclosed study related to the same Phase Ib/IIa study (BP22572) which included both Kadcyla and Perjeta.
The trial in question was predominantly a Phase Ib, multi-centre, open-label study to assess the feasibility of Kadcyla plus docetaxel in patients with HER2-positive metastatic breast cancer, and Kadcyla plus docetaxel with or without Perjeta, in patients with HER2-positive locally advanced breast cancer. There was also a Phase IIa component to obtain further safety and efficacy data from the maximum tolerated dose from each patient cohort. The trial was initiated, led and conducted through Roche’s global organisation. It was conducted in multiple sites globally and there was one UK trial site. Accordingly, Roche stated that the study fell within the scope of the Code with regard to disclosure.
The trial was completed (last patient, last visit) on 24 October 2013, after the date of commercialisation for both Kadcyla and Perjeta. As a result, having considered the Joint Positions 2008 and 2009, Roche stated that this date should be the reference point for disclosure timeframes thereafter.
From the Decision Tree developed in the context of previous complaints Roche submitted that as Perjeta was first licensed in June 2012 it should be considered within the scope of the 2012 Code which referred solely to the Joint Position 2008. As Kadcyla was first licensed in February 2013 it should be considered within the scope of the Second 2012 Edition of the Code which referred to the Joint Position 2009.
Roche stated that as the BP22572 trial completed on 24 October 2013, after the first approval dates for both Kadcyla and Perjeta, the timelines for posting the trial results on Clinicaltrials.gov should have been one year after study completion, ie by 24 October 2014.
BP22572 was registered on clinicaltrials.gov on 6 July 2009 (NCT00934856) however due to an incorrect Phase I categorisation (rather than Phase I/II) within Roche, results were not posted to Clinicaltrials.gov. (Phase I trials were excluded from the registration and results submission requirements of FDAAA 801). The trial had now been reclassified within Roche and the Clinicaltrials.gov posting of results was in progress.
Although trials could be registered on EudraCT, the results section was not launched until 21 July 2014. For any interventional clinical trials that ended on or after 21 July 2014, it was compulsory for sponsors to post results within six or twelve months following the end of the trial, depending on the type of trial concerned. For other trials (where regulated by Directive 2001/20/EC) that ended <1 year prior to the finalisation of the programming (21 July 2014) the ‘Trial results: modalities and timing of posting’, document on the EudraCT website stated that results should be posted ≤12 months after finalisation of the programming.
As BP22572 completed on 24 Oct 2013, the results were due to be posted on EudraCT by 21 July 2015. Roche initiated the process of posting this information in March 2015 however due to issues with the third party vendor supporting the submission and review/approval delays, the deadline of 21 July 2015 was missed.
The EudraCT system was then withdrawn from 31 July 2015 until 13 January 2016 as stated in the release notes on the EudraCT website. Results for BP22572 were posted by Roche on 17 February 2016 and following validation by the EMA they were finalised on the system on 4 March 2016. The results publication on EudraCT was removed for a period of time in 2016 stating that ‘the results have been removed from public view whilst they are reviewed and may need to be corrected before being returned to public view’. Roche was not made aware of this by the EMA and the results were returned to public view following an enquiry from Roche to the EMA.
Disclosure in the scientific literature
Roche stated that the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature 2010 was first referenced within the Second 2012 Edition of the Code. Strictly speaking therefore it only applied to Kadcyla in the context of this complaint however Roche recognised the need for adherence to the Joint Position for Perjeta also regardless of when the ABPI Code applied to it.
The requirement in this Joint Position stated that results of completed industry-sponsored clinical trials should be submitted for publication wherever possible within 12 months and no later than 18 months of the completion of clinical trials (for already marketed medicinal products) (therefore in this case, by April 2015). A primary manuscript was submitted to The Journal of Clinical Oncology on 15 May 2015. This manuscript was rejected and re-submitted to Annals of Oncology and after two rounds of time consuming peer-reviewed comments it was subsequently published on 6 April 2016.
Data from the BP22572 trial was published at the San Antonio Breast Cancer Symposium, in December 2012 (interim analysis) and December 2013 (data from locally advanced breast cancer (LABC) patients treated at the maximum tolerated dose).
Summary
Roche submitted that the requirement in the ABPI Code was based around the disclosure of clinical trials rather than by product. Roche appreciated that the PMCPA did not have the full detail regarding the trials associated with each product and thus raised two separate complaints. It submitted that as both complaints related to the same trial they should be combined and assessed as one complaint rather than two.
Roche stated that with regard to trial BP22572, it accepted that it did not disclose details of this trial in accordance with the requirements of Clause 13.1 set out in the relevant ABPI Code detailed above. In failing to disclose details of this trial in line with disclosure requirements, Roche also accepted its failure to maintain high standards at all times.
Roche stated that whilst it was unfortunate that this trial was not disclosed within the required timeframes, it had since been published both on the EudraCT platform and within the scientific literature ensuring full disclosure. Furthermore Roche did not believe that the delay in disclosure of this trial would have impacted patient safety and/or public health.
Roche stated that it took its commitment to disclosure very seriously and strove to operate within clearly documented processes and procedures. In addition, Roche had recently implemented a new clinical trial disclosure internal review platform which would be used to manage the process of clinical trial protocol and results disclosure to public registries. This would improve its oversight of the trials to be processed, with timelines and deliverables built and automated within the system.
Roche regretted that the trial at issue was not disclosed within the required timeframes however Roche submitted that a breach of Clause 2, a sign of particular censure, was not warranted in this case.
GENERAL COMMENTS FROM THE PANEL
The Panel noted that all the cases would be considered under the Constitution and Procedure in the 2016 Code as this was in operation when the CMRO study was published and the complaint proceedings commenced. The Panel noted that the study concluded that of the completed trials associated with 34 new medicines licensed to 24 different companies in 2013, results of 90% had been disclosed within 12 months and results of 93% had been disclosed by 31 July 2015.
The Panel noted that the CMRO publication in question was an extension of previously reported data from two studies, one related to new medicines approved in Europe in 2009, 2010 and 2011 (Rawal and Deane 2014) which found that over threequarters of all these trials were disclosed within 12 months and almost 90% were disclosed by the end of the study. That study was the subject of an external complaint which gave rise to 27 cases in 2013 and 2014. The second study (Rawal and Deane 2015) was not the subject of external complaint but was taken up under Paragraph 5.1 of the Constitution and Procedure in 2015 leading to 15 cases. The second study found that the results of 90% had been disclosed within 12 months and results of 92% had been disclosed by 31 July 2014. Most of these cases were not in breach of the Code because they were not within the scope of the Code as there was no UK involvement and therefore only limited details were published on the PMCPA website. The present case was not the subject of external complaint. The study itself formed the basis of the complaint.
The Panel considered that the first issue to be determined was whether the matter was covered by the ABPI Code. If the research was conducted on behalf of a UK pharmaceutical company (whether directly or via a third party) then it would be covered by the ABPI Code. If a trial was run by a non UK company but had UK involvement such as centres, investigators, patients etc it was likely that the Code would apply. The Panel appreciated the global nature of much pharmaceutical company sponsored clinical research and a company located in the UK might not be involved in research that came within the ABPI Code. It was a well established principle that UK pharmaceutical companies were responsible for the activities of overseas affiliates if such activities came within the scope of the Code such as activities relating to UK health professionals or activities carried out in the UK.
Clause 13.1 of the 2016 and 2015 editions of the Code stated that companies must disclose details of clinical trials in accordance with the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature.
The relevant supplementary information stated that this clause required the provision of details about ongoing clinical trials (which must be registered within 21 days of initiation of patient enrolment) and the results of completed trials for medicines licensed for use and commercially available in at least one country. Further information was to be found in the current Joint Position on the Disclosure of Clinical Trial Information via Clinical
Trial Registries and Databases and the current Joint Position on the Publication of Clinical Trial Results in the Scientific Literature, both at www.ifpma.org. en/ethics/clinical-trials-disclosure.html. Companies must include on the home page of their website, information as to where details of their clinical trials could be found.
The Panel noted that the first Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases was agreed in 2005 by the International Federation of Pharmaceutical Manufacturers and Associations (IFPMA), the European Federation of Pharmaceutical Industries and Associations (EFPIA), the Japanese Pharmaceutical Manufacturers Association (JPMA) and the Pharmaceutical Research and Manufacturers of America (PhRMA). The announcement was dated 6 January 2005.
The Panel noted that Article 9, Clinical Research and Transparency, of the most recent update of the IFPMA Code of Practice (which came into operation on 1 September 2012) included a statement that companies disclose clinical trial information as set out in the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases (2009) and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature (2010). As companies had, in effect, agreed the joint positions their inclusion in the IFPMA Code should not have made a difference in practice to IFPMA member companies but meant that IFPMA member associations had to amend their codes to reflect Article 9. Pharmaceutical companies that were members of national associations but not of IFPMA would have additional disclosure obligations once the national association amended its code to meet IFPMA requirements. The disclosures set out in the joint positions were not required by the EFPIA Codes.
The Panel noted that even if the UK Code did not apply many of the companies listed in the study were members of IFPMA and/or EFPIA.
The Panel considered that it was good practice for clinical trial results to be disclosed for medicines which were first approved and commercially available after 6 January 2005 (the date of the first joint position). This was not necessarily a requirement of the ABPI Codes from that date as set out below.
As far as the ABPI Code was concerned, the Panel noted that the first relevant mention of the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases 2005 was in the supplementary information to Clause 7.5 of the 2006 Code:
‘Clause 7.5 Data from Clinical Trials
Companies must provide substantiation following a request for it, as set out in Clause 7.5. In addition, when data from clinical trials is used companies must ensure that where necessary that data has been registered in accordance with the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases 2005.’
Clause 7.5 of the 2006 Code required that substantiation be provided at the request of health professionals or appropriate administrative staff. Substantiation of the validity of indications approved in the marketing authorization was not required. The Panel considered this was not relevant to the complaint being considered which was about disclosure of clinical trial results. The Joint Position 2005 was mentioned in the supplementary information to Clause 21.5 but this did not relate to any Code requirement to disclose clinical trial results.
In the 2008 ABPI Code (which superceded the 2006 Code and came into operation on 1 July 2008 with a transition period until 31 October 2008 for newly introduced requirements), Clause 21 referred to scientific services and Clause 21.3 stated: ‘Companies must disclose details of clinical trials.’
The relevant supplementary information stated:
‘Clause 21.3 Details of Clinical Trials
This clause requires the provision of details about ongoing clinical trials (which must be registered within 21 days of initiation of patients enrolment) and completed trials for medicines licensed for use in at least one country. Further information can be found in the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases 2005 (http:// clinicaltrials.ifpma.org).
Details about clinical trials must be limited to factual and non-promotional information. Such information must not constitute promotion to health professionals, appropriate administrative staff or the public.’
In the 2011 Code (which superceded the 2008 Code and came into operation on 1 January 2011 with a transition period until 30 April 2011 for newly introduced requirements), the supplementary information to Clause 21.3 was updated to refer to the 2008 IFPMA Joint Position.
In the Second 2012 Edition (which came into operation on 1 July 2012 with a transition period until 31 October 2012 for newly introduced requirements), changes were made to update the references to the joint position and to include the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature. Clause 21.3 now stated:
‘Companies must disclose details of clinical trials in accordance with the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature.’
The relevant supplementary information stated:
‘Clause 21.3 Details of Clinical Trials
This clause requires the provision of details about ongoing clinical trials (which must be registered within 21 days of initiation of patients enrolment) and completed trials for medicines licensed for use in at least one country. Further information can be found in the Joint Position on the Disclosure of Clinical Trial Information via Clinical
Trial Registries and Databases 2009 and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature 2010, both at http:// clinicaltrials.ifpma.org.
Details about clinical trials must be limited to factual and non-promotional information. Such information must not constitute promotion to health professionals, appropriate administrative staff or the public.’
The Panel noted that in the 2014 ABPI Code the disclosure requirements which had previously been stated in Clause 21 had been moved to Clause 13. In addition, the supplementary information stated that companies must include on their website information as to where details of their clinical trials could be found. The 2014 Code came into effect on 1 May 2014 for newly introduced requirements following a transition period from 1 January 2014 until 30 April 2014. These requirements were to be found in Clause 13.1 of the 2015 Code. The relevant supplementary information had been amended in the 2015 Code to replace the year of the relevant joint positions with the word ‘current’, to add a reference to the medicine being licensed and ‘commercially available’ and to update the website address. The 2015 Code came into effect on 1 May 2015 for newly introduced requirements following a transition period from 1 January 2015 until 30 April 2015. Similarly the 2016 Code came into effect on 1 May 2016 for newly introduced requirements following a transition from 1 January 2016 to 30 April 2016. The study at issue was posted online on 25 November 2016.
The Panel examined the Joint Position on the Disclosure of Clinical Trial Information which was updated on 10 November 2009 and superseded the Joint Position 2008. With regard to clinical trial registries the document stated that all trials involving human subjects for Phase I and beyond at a minimum should be listed. The details should be posted no later than 21 days after the initiation of enrolment. The details should be posted on a free, publicly accessible, internet-based registry. Examples were given. Each trial should be given a unique identifier to assist in tracking. The Joint Position 2009 provided a list of information that should be provided and referred to the minimum Trial Registration Data Set published by the World Health Organisation (WHO). The Joint Position 2009 referred to possible competitive sensitivity in relation to certain data elements and that, in exceptional circumstances, this could delay disclosure at the latest until after the medicinal product was first approved in any country for the indication being studied. Examples were given.
The Panel noted that the matter for consideration related to the disclosure of clinical trial results.
With regard to the disclosure of clinical trial results the Joint Position 2009 stated that the results for a medicine that had been approved for marketing and was commercially available in at least one country should be publicly disclosed. The results should be posted no later than one year after the medicine was first approved and commercially available. The results for trials completed after approval should be posted one year after trial completion – an adjustment to this schedule was possible to comply with national laws or regulations or to avoid compromising publication in a peer-reviewed medical journal.
The Joint Position 2009 included a section on implementation dates and the need for companies to establish a verification process.
The Joint Position 2005 stated that the results should be disclosed of all clinical trials other than exploratory trials conducted on a medicine that was approved for marketing and was commercially available in at least one country. The results generally should be posted within one year after the medicine was first approved and commercially available unless such posting would compromise publication in a peer reviewed medical journal or contravene national laws or regulations. The Joint Position 2008 was dated 18 November 2008 and stated that it superseded the Joint Position 2005 (6 January and 5 September). The Joint Position 2008 stated that results should be posted no later than one year after the product was first approved and commercially available in any country. For trials completed after initial approval these results should be posted no later than one year after trial completion. These schedules would be subject to adjustment to comply with national laws or regulations or to avoid compromising publication in a peer reviewed medical journal.
The Joint Position on the Publication of Clinical Trial Results in the Scientific Literature was announced on 10 June 2010. It stated that all industry sponsored clinical trials should be considered for publication and at a minimum results from all Phase III clinical trials and any clinical trials results of significant medical importance should be submitted for publication. The results of completed trials should be submitted for publication wherever possible within 12 months and no later than 18 months of the completion of clinical trials for already marketed medicines and in the case of investigational medicines the regulatory approval of the new medicine or the decision to discontinue development.
Having examined the various codes and joint positions, the Panel noted that the Joint Position 2005 excluded any clinical trials completed before 6 January 2005. The position changed on 18 November 2008 as the Joint Position 2008 did not have any exclusion relating solely to the date the trial completed. The Joint Position 2009 was similar to the Joint Position 2008 in this regard.
The Panel noted that deciding which Code, and thus which joint position applied, was complicated. It noted that the 2011 Code which, taking account of the transition period, came into operation on 1 May 2011, was the first edition of the Code to refer to the Joint Position 2008.
The Panel concluded that from 1 November 2008, (allowing for the transition period) until 30 April 2011 under the 2008 Code companies were required to follow the Joint Position 2005. From 1 May 2011 until 30 April 2012 under the 2011 Code and 1 May 2012 until 31 October 2012 under the 2012 Code companies were required to follow the Joint Position 2008. Since 1 November 2012 companies were required to follow the Joint Position 2009. The Panel considered that since the 2008 Code companies were, in effect, required to comply with the joint position cited in the relevant supplementary information. The relevant supplementary information gave details of what was meant by Clause 21.3 (Clause 13.1 in the 2014, 2015 and 2016 Codes). The Panel accepted that the position was clearer in the Second 2012 Edition of the Code. The Panel noted that the 2011 Code should have been updated to refer to the Joint Position 2009.
For medicines first licensed and commercially available in any country from 1 November 2008 until 30 April 2011 the results of clinical trials completed before 6 January 2005 would not have to be posted.
From 1 May 2011 there was no exclusion of trials based solely on completion date and so for a product first licensed and commercially available anywhere in the world after 1 May 2011 the applicable joint positions required relevant clinical trial results to be posted within a year of the product being first approved and commercially available or within a year of trial completion for trials completed after the medicine was first available.
Noting that the CMRO study referred to licensed products the Panel considered that the trigger for disclosure was the date the product was first approved and commercially available anywhere in the world. This would determine which version of the Code (and joint position) applied for trials completed prior to first approval. The next consideration was whether the trial completed before or after this date. For trials completing after the date of first approval, the completion date of the trial would determine which Code applied. The Panel considered that the joint positions encouraged disclosure as soon as possible and by no later than one year after first availability or trial completion as explained above. The Panel thus considered that its approach was a fair one. In this regard, it noted that the matter for consideration was whether or not trial results had been disclosed, all the joint positions referred to disclosure within a one year timeframe and companies needed time to prepare for disclosure of results. The Panel considered that the position concerning unlicensed indications or presentations of otherwise licensed medicines etc would have to be considered on a case by case basis bearing in mind the requirements of the relevant joint position and the legitimate need for companies to protect intellectual property rights.
The Panel referred to the decision tree in the previous cases (for example Case AUTH/2654/11/13 et al) which had been updated in 2015 and published in Case AUTH/2763/5/15. The Panel updated the 2015 decision tree to include the 2016 Code.
Decision Tree
Updated Decision tree developed by the Panel
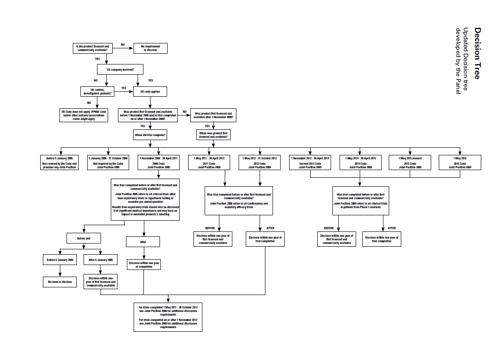
The Panel considered that companies would be well advised to ensure that all the clinical trial results were disclosed as required by the codes and joint positions. The Panel considered that there was no complaint about whether the results disclosed met the requirements of the joint positions so this was not considered. In the Panel’s view the CMRO publication at issue and thus the matter for consideration was only about whether or not trial results had been disclosed and the timeframe for such disclosure. The CMRO publication focussed on the disclosure of evaluable trial results and the Panel only considered those evaluable trials.
The Panel noted that its consideration of these cases relied upon the information provided by the respondent companies. The CMRO publication did not identify the studies evaluated; it only provided quantitative data. The Panel noted that the study related to products approved for marketing by the EMA in 2013 and searched for the data between 1 May and 31 July 2015. The study was published online on 25 November 2016. It appeared that the authors of the CMRO publication had contacted various companies for additional information.
The Panel noted that the date the product was first licensed and commercially available anywhere in the world might pre-date EMA approval.
PANEL RULING
The Panel noted the CMRO publication in that one evaluable trial had not been disclosed within the timeframe for Kadcyla. The disclosure percentage at 12 months measured from the later of the first date of regulatory approval or trial completion date was 91%. The disclosure percentage at 31 July 2015 was 91%.
The Panel noted Roche’s submission that Kadcyla was first approved and commercially available in in the US on 22 February 2013.
The Panel noted the CMRO publication in that one evaluable trial for Perjeta had not been disclosed within the timeframe. The disclosure percentage at 12 months was 95%. The disclosure percentage at 31 July 2015 was 100%.
The Panel noted Roche’s submission that Perjeta was first approved and commercially available in the US on 8 June 2012.
The Panel noted Roche’s submission that in both of the instances above, the single alleged undisclosed trial related to the same Phase Ib/IIa study (BP22572) which included both Kadcyla and Perjeta. The trial was initiated, led and conducted through Roche’s global organisation; it was conducted in multiple sites globally and included one UK trial site and thus fell within the scope of the ABPI Code with regard to disclosure as acknowledged by Roche.
In the circumstances, the Panel considered that the most recent applicable Code and Joint Position would apply ie the Second 2012 Code and the Joint Position 2009 based on the first commercialisation of Kadcyla.
The trial was completed (last patient, last visit) on 24 October 2013. This completion date was after the date of commercialisation for both Kadcyla and Perjeta. The Panel noted that on the information before it the trial results should have been posted on a publicly accessible, internetbased clinical trials database by 24 October 2014. The Panel noted Roche’s submission that the trial at issue was registered on ClinicalTrials.gov on 6 July 2009 (NCT00934856) however due to an incorrect Phase I categorisation (rather than Phase I/II) within Roche, results were not posted to ClinicalTrials.gov. (Phase I trials were excluded from the registration and results submission requirements of FDAAA 801). The trial had now been reclassified within Roche.
The Panel noted that data from the BP22572 trial was published at the San Antonio Breast Cancer Symposium, in December 2012 (interim analysis) and December 2013 (data from LABC patients treated at the maximum tolerated dose), however the complete results had not been posted on a publicly accessible, internet based, clinical trials database within the required timeframe as acknowledged by Roche. The Panel thus ruled a breach of Clause 13.1. The delay in disclosure meant that high standards had not been maintained and a breach of Clause 9.1 was ruled.
As the data had now been disclosed the Panel considered that there was no breach of Clause 2 and ruled accordingly.
Complaint received 29 November 2016
Cases completed 13 March 2017
see Cases: 3005,2908,2906,2903,2898,2763,2676,2674,2673,2672,2671,2670,2669,2667,2666,2665,2664,
2663,2662,2661,2659,2657,2654