ANONYMOUS CONTACTABLE MEMBER OF THE PUBLIC v ASTRAZENECA
NO BREACH OF THE CODE
Clinical trial disclosure (Iressa)
An anonymous, contactable member of the public complained about the information published as ‘Clinical Trial Transparency: an assessment of the disclosure results of company-sponsored trials associated with new medicines approved recently in Europe’. The study was published in Current Medical Research & Opinion (CMRO) on 11 November 2013.
The study authors were Dr B Rawal, Research, Medical and Innovation Director at the ABPI and B R Deane, a freelance consultant in pharmaceutical marketing and communications. Publication support for the study was funded by the ABPI.
The study surveyed various publicly available information sources for clinical trial registration and disclosure of results searched from 27 December 2012 to 31 January 2013. It covered 53 new medicines (except vaccines and fixed dose combinations) approved for marketing by 34 companies by the European Medicines Agency (EMA) in 2009, 2010 and 2011. It included all completed company-sponsored clinical trials conducted in patients and recorded on a clinical trial registry and/or included in a European Public Assessment Report (EPAR). The CMRO publication did not include the specific data for each product. This was available via a website link and was referred to by the complainant. The study did not aim to assess the content of disclosure against any specific requirements.
The complainant stated that the study detailed a number of companies which had not disclosed their clinical trial results in line with the ABPI for licensed products. The complainant provided a link to relevant information which included the published study plus detailed information for each product that was assessed.
The summary output for each medicine set out the sources for all trials found, irrespective of sponsor and an analysis of publication disclosure in the form of a table which gave details for the studies for Iressa (gefitinib).
The detailed response from AstraZeneca is given below.
General detailed comments from the Panel are given below.
The Panel noted the CMRO publication in that twenty-nine Iressa studies had not been disclosed in the timeframe. The disclosure percentage was 56%. Twelve studies had not been disclosed giving a disclosure percentage at 31 January 2013 for trials completed at 31 January 2012 of 84%. A footnote stated that the majority of Phase II/III trials were completed prior to FDAAA 801 requirements. The remaining undisclosed trials were in the process of publication.
The Panel noted AstraZeneca’s submission regarding the studies. Iressa was first approved and commercially available in Japan in 2002.
The Panel noted that of the remaining 38 trials (53 minus 15 investigator-sponsored trials), 35 were Phase I, exploratory Phase II or Phase III studies all of which completed before 1 November 2008. In that regard, there was no requirement under the Code to disclose these studies. The Panel thus ruled no breach of the 2008 Code including Clause 2.
An AstraZeneca Thailand non-interventional study completed in August 2010, which was after Iressa was first approved and commercially available. The Panel noted AstraZeneca’s submission that these results were disclosed on its own website in November 2010. It was not clear whether there was any UK involvement and the Joint Position 2005 appeared not to require disclosure of the results of a non interventional trial. In any event the results had been disclosed publicly within one year and thus the Panel ruled no breach of the 2008 Code including Clause 2.
The Panel noted that the results from two trials remained undisclosed – an AstraZeneca Canada study which completed in August 2011 and an AstraZeneca Taiwan study which completed in August 2009. AstraZeneca submitted that the publication of the results was expected.
The Panel considered that although AstraZeneca was a UK registered company, the company’s arrangements were such that it was clear that the responsibility for disclosure was with the local company. It considered that the matter was potentially covered by the UK Code but as the responsibilities had been made very clear in a company standard operating procedure it ruled no breach of the 2008 Code including Clause 2 in relation to the AstraZeneca Taiwan trial and no breach of the 2011 Code including Clause 2 in relation to the AstraZeneca Canada study.
An anonymous, contactable member of the public complained about the information published as ‘Clinical Trial Transparency: an assessment of the disclosure results of company-sponsored trials associated with new medicines approved recently in Europe’. The study was published in Current Medical Research & Opinion (CMRO) on 11 November 2013.
The study authors were Dr B Rawal, Research, Medical and Innovation Director at the ABPI and B R Deane, a freelance consultant in pharmaceutical marketing and communications. Publication support for the study was funded by the ABPI.
The study surveyed various publicly available information sources for clinical trial registration and disclosure of results searched from 27 December 2012 to 31 January 2013. It covered 53 new medicines (except vaccines and fixed dose combinations) approved for marketing by 34 companies by the European Medicines Agency (EMA) in 2009, 2010 and 2011. It included all completed company-sponsored clinical trials conducted in patients and recorded on a clinical trial registry and/or included in a European Public Assessment Report (EPAR). The CMRO publication did not include the specific data for each product. This was available via a website link and was referred to by the complainant. The study did not aim to assess the content of disclosure against any specific requirements.
COMPLAINT
The complainant stated that the study detailed a number of companies which had not disclosed their clinical trial results in line with the ABPI for licensed products. The complainant provided a link to relevant information which included the published study plus detailed information for each product that was assessed.
The summary output for each medicine set out the sources for all trials found, irrespective of sponsor and an analysis of publication disclosure in the form of a table which gave details for the studies for each product.
The data for Iressa (gefitinib) were as follows:
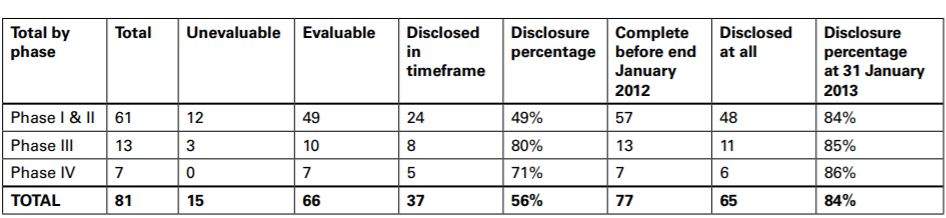

The explanation of terms given in the documentation was as follows:
Total
Total number of trials identified which were completed and/or with results disclosed
Unevaluable
Trials within the total which could not be evaluated (due to either trial completion date or publication date being missing or unclear) – excluded from the analysis
Evaluable
Trials with all criteria present including dates, and hence the base which could be evaluated for the assessment
Results disclosed in timeframe
Evaluable trials which fully complied with publication requirements, ie summary results disclosed (in registry or journal) within 12 months of either first regulatory approval date or trial completion date, whichever was later
Disclosure percentage
Proportion of evaluable trials which were fully disclosed
Completed before end of January 2012
Number of studies completed before end January 2012 (or already disclosed)
Results disclosed at all
Number of trials with any publication of results at any time
Disclosure percentage at 31 January 2013
Proportion of trials completed by end January 2012 which were now disclosed
* * *
The complainant listed the companies he/she would like to complain about and this included AstraZeneca.
The complainant alleged that all of the companies listed had breached Clauses 2, 9 and 21 of the Code.
When writing to AstraZeneca, the Authority drew attention to Clauses 1.8 and 21.3 of the Second 2012 Edition of the Code and noted that previous versions of the Code might also be relevant.
RESPONSE
AstraZeneca stated that it took its compliance with pharmaceutical industry codes of practice and all underlying legislation and regulations very seriously.
General points on disclosure
AstraZeneca had a long-standing commitment to make information about its clinical research publicly available to enhance the scientific understanding of how its medicines worked and in the medical interest of patients. As a company, its disclosure policies went above and beyond the current legal mandated requirements.
AstraZeneca’s investigational clinical trials were registered on the US National Library of Medicine’s website before the first patient was enrolled and to other websites within timelines as required by law. Additionally, it posted basic information on the company website which was publicly accessible.
AstraZeneca considered that although transparency of clinical trial results and applicable information from its investigational clinical trials contributed to public confidence in medicines and improved public health and scientific knowledge, it recognised that increased transparency, in both the reactive and proactive disclosure contexts, must be balanced with the legally required protection of personal data, intellectual property and confidential information.
Thus AstraZeneca was committed to communicating accurate and meaningful information about its sponsored clinical trials in a timely, accurate, balanced and complete manner, regardless of outcome. AstraZeneca’s current and planned clinical trials transparency position met or exceeded all existing legally required and regulatory standards. AstraZeneca submitted that it:
- registered and posted results from all of its Phase I-IV interventional trials, including healthy volunteer trials, on ClinicalTrials.gov and other applicable legally required websites, as well as its own website
- registered non-interventional studies and disclosed the results of trials conducted on marketed products on any and all legally required websites in addition to its own website posted trial results, synopses and other information on its website for products approved in countries that did not legally require disclosure
- The timelines of disclosure were as follows:
– Results of trials with already marketed medicines were posted within one year of completion. Results of trials with medicines in development were posted within 30 days of first regulatory approval for the new medicine where trials had completed at least one year. When a medicine in development was discontinued, results were published within one year of the public announcement of the decision, unless analysis and interpretation of the data were not sufficiently complete, in which case an explanation for the delay was posted together with the anticipated date when the results would be posted.
– For marketed medicines and recently approved medicines where AstraZeneca considered there to be good cause to delay posting of results, it sought the necessary approval according to applicable law. Where approved, it posted an explanation for the delay and the anticipated date when the results would be posted.
In essence, AstraZeneca posted the results of all of its clinical trials in all stages of clinical development on several public websites – regardless of outcome (positive or negative) – and included products which had been discontinued in development.
Comments on the complaint
AstraZeneca stated that the purpose of the CMRO study was to identify from the cohort of all completed company sponsored clinical trials, carried out in patients and relating to new medicines approved by the EMA in 2009, 2010 and 2011, studies for which results were not posted in a ‘timely manner’; in other words and according to the protocol, studies identified through searching clinical trial registries and/or included in a European Public Assessment Report (EPAR) for which results had not been disclosed, either within twelve months of the later of either first regulatory approval or trial completion, or by 31 January 2013.
The complainant specifically referred to Iressa, which was first launched in Japan in 2002, followed by the US in 2003. The FDA subsequently updated the conditional approval indication in 2005 to exclude new patients following failure of the ISEL study to demonstrate extended survival and AstraZeneca subsequently withdrew the NDA in 2011. Following further research to identify the patient population most benefiting, the EMA approved Iressa for patients with EGFR mutation positive Non-Small Cell Lung Cancer (NSCLC) in 2009 based on the results of the IPASS study. A list of the worldwide marketing authorizations for Iressa was provided and was accurate when collated in October 2013.
AstraZeneca submitted that the scope of information requested by the case preparation manager was unreasonable, in that it went beyond the basis of the complaint which specifically referred to the CMRO publication. Therefore AstraZeneca had completed an in-depth response to the allegation that it had failed to disclose results according to requirements for clinical trials for the studies included within the CMRO publication, and had not responded to the broader request for a specific listing of all ongoing and completed Iressa clinical trials and the information pertaining to these trials. In addition, it had only completed the request for UK specific information for those trials where the company considered the information would inform the PMCPA with regard to the complaint and its scope. AstraZeneca had taken this approach in the interests of responding within PMCPA timelines.
AstraZeneca was asked to consider the requirements of Clause 21.3 in its response. Clause 21.3 (2008) required the posting of information about ongoing and completed clinical trials and referred to the 2005 IFPMA Joint Position on Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases. The only difference in Clause 21.3 (2011), as written, was recognition of the 2008 IFPMA Joint Position on Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases as the core reference. AstraZeneca submitted that the principal difference between the two joint positions was the commitment to publish exploratory as well as confirmatory trials.
Summary
AstraZeneca submitted that it was not in breach of Clause 21.3, in that each of the Iressa clinical studies completed before the requirements of the Code or they fell outside its jurisdiction, as no UK patient, site, investigator, or UK-based AstraZeneca member of staff was involved.
In addition, the data below showed that many of the trials had been published in journals and those publications were listed on the Clinical Trials.gov as provided by US National Institutes of Health, or on the US National Library of Medicine National Institutes of Health, and for those which had not yet been published, AstraZeneca remained committed to posting/publication of results, as stated in the publication that formed the basis of this complaint. This was in line with the AstraZeneca disclosure position.
Investigation and findings
Following in-depth analysis of the publicly available information on the identified Iressa studies, the researchers gave AstraZeneca a list of studies that, in their opinion, failed to meet the requirements of the protocol. This list of 53 clinical trials was the principal basis upon which AstraZeneca investigated and responded to this complaint. A copy was provided. This number was higher than the highest assessment of undisclosed trials discovered by the researchers (n=44; total number of studies identified (n=81) minus those considered both evaluable and disclosed within timelines (n=37)); this was the most reliable and least conservative information regarding the clinical trials relevant to this complaint that AstraZeneca had. However, AstraZeneca confirmed that during an in-depth internal review of Iressa clinical trials, it had not discovered any other trials that fell within the scope of the Food and Drug Administration Amendments Act of 2007 Section 801, and/or any other applicable requirements, including of Clause 21.3 of the Code (2008 and 2011) and all applicable company policy requirements and where results were not disclosed accordingly.
A spreadsheet set out the data for each of the 53 trials listed. In summary:
- Fifteen of the studies identified by the researchers as potentially being out of compliance with the protocol were investigator sponsored studies, and therefore accountability for disclosure/publication of those results was with the sponsor of the study, not AstraZeneca. Consequently they fell out with the requirements of Clause 21.3 of the Code.
- Thirty-one of the studies identified by the researchers as potentially being out of compliance with the protocol were either Phase I or exploratory Phase II studies completed before the cut-off date of 6-months before publication of the 2008 IFPMA Joint Position; therefore they fell out with the requirements of disclosure/publication of this type of study, and consequently also Clause 21.3 of the ABPI Code (2011).
– For 23 of the studies all results had since been disclosed, on clinicaltrials.gov and/or AstraZenecaClinicalTrials.com (Section 2.1).
– The results from eight of the studies had not been published, though AstraZeneca remained committed to ensuring their publication over time (Section 2.2).
- Four of the studies identified by the researchers as potentially being out of compliance with the protocol were Phase III studies that were completed before the publication date of the 2005 IFPMA Joint Position; therefore they fell outwith the requirements of publication of this type of study and consequently Clause 21.3 of the ABPI Code (2008 and 2011).
– Two studies had not had results disclosed, though AstraZeneca remained committed to ensuring their publication over time.
– The results from two studies had since been disclosed.
- One study was a local phase IV non-interventional study, sponsored by AstraZeneca Taiwan, which completed in August 2010. This study was identified incorrectly as being potentially out of compliance with the protocol, as it was not an interventional study and results were in fact disclosed on AstraZenecaClinicalTrials.com in November 2010. A summary of the trial from the AstraZeneca website was provided.
- Of the remaining two studies, AstraZeneca UK recognised that the studies did not report results within the timelines required by the IFPMA Joint Position on Disclosure of Clinical
Trial Information via Clinical Trial Registries and Databases (2008); both studies were local studies, conducted overseas, with no UK patient, site, investigator and were both outwith the control or responsibility of the UK affiliate, or indeed any study team based within the UK.
– One study was originally started as a local phase II investigator sponsored study in Canada, and completed in August 2011.
This study was listed on clinicaltrials.gov as AstraZeneca sponsored, in error, and any and all postings and/or disclosure of applicable information would be the responsibility of the investigator who initiated the study. To mitigate disclosure implications which led to a delay in disclosure of results, AstraZeneca Canada was working closely with the responsible investigator to ensure disclosure of results.
- The final study was a local phase IV study, again sponsored by AstraZeneca Taiwan, which was terminated in August 2009, with only 14 patients recruited and a safety summary produced.
– The local study team in AstraZeneca Taiwan was currently expediting disclosure of these limited results on clintrials.gov.
Conclusion
AstraZeneca submitted as was evident from the information supplied above, each of the AstraZeneca Iressa clinical studies identified in the research which formed the basis of the complaint:
- was outside the legal requirements under Food & Drug Administration Amendments Act of 2007, (FDAAA, 2007) and/or
- had results reported on clinicaltrials.gov and/or the AstraZeneca website
- was in the process of being published.
In addition, many had also been published in journals and those publications were listed on the Clinical Trials.gov as provided by US National Institutes of Health, or on the US National Library of Medicine National Institutes of Health. For those that had not yet been published, AstraZeneca was committed to posting/publication of results, as stated in the report that forms the basis of this complaint and in line with the company disclosure position, as stated above.
AstraZeneca submitted that it was not in breach of Clause 21.3 (2008 or 2011), as the studies identified by the researchers as being out of compliance with their protocol, either fell outwith the requirements of the Code, in that their completion predated the requirements of the Code, or they fell outwith the jurisdiction of the Code as there was no UK involvement.
Subsequent to completion of the principal draft of its response, AstraZeneca was sent a spreadsheet detailing all trials identified by the researchers using the publication search protocol. AstraZeneca highlighted the trials where disclosure status was queried, to aid the PMCPA in cross-referencing.
In response to a request for additional information AstraZeneca stated that the two studies detailed as not reported within the joint position timeframe did not involve any UK team from within the Global AstraZeneca organisation. Accountability for the delivery of the study sat with the local study delivery team within the country (Canada and Taiwan, in this case). Responsibility for registration of the study and for the posting and publication of results sat with the local study team leader and accountability with the local director or vice president, medical.
AstraZeneca provided copies of SOPs that referred to clinical trial results and where the responsibility for disclosure sat. The current SOP was provided. The versions valid in 2009 and 2011 were not found, however, AstraZeneca submitted that there was no significant difference in process, roles and responsibilities between current and past versions.
AstraZeneca stated that the Clinical Trials Disclosures Procedures and Responsibilities document detailed the accountability of the marketing company (affiliate) medical director and the responsibility of the study team leader – namely to complete the required templates and submit them to the clinical trials transparency (CTT) team. This team then ensured that the documentation was checked by all the necessary central teams and posted on the appropriate websites. AstraZeneca stated that the responsibility and accountability for clinical trial registration and results posting sat clearly with the local study team who initiated the process by completing and submitting the templates in a timely fashion and ensured the accuracy and completeness of the submitted information; not the CTT team, whose responsibility, though important for compliance monitoring and tracking, was primarily administrative. This team was currently based in Poland, and was previously a US based team.
Iressa was first licensed in Japan in 2002. Iressa 250mg once daily originally received approval on 5 July, 2002. It was originally licensed for the treatment of inoperable or recurrent non-small cell lung cancer (NSCLC) in Japan, whilst the European licence, granted in 2009, was for EGFR mutation positive NSCLC; the broader indication was never granted in the EU.
GENERAL COMMENTS FROM THE PANEL
The Panel noted the ABPI involvement in the study. However, a complaint had been received and it needed to be considered in the usual way in line with the PMCPA Constitution and Procedure. The Panel noted that all the cases would be considered under the Constitution and Procedure in the Second 2012 Edition as this was in operation when the complaint was received. The addendum (1 July 2013 which came into effect on 1 November 2013) to this Code only related to Clause 16 and was not relevant to the consideration of these cases.
The Panel noted that the study concluded that the results of over three quarters of all company sponsored clinical trials were disclosed within a year of completion or regulatory approval and almost 90% were disclosed by 31 January 2013 which suggested transparency was now better than had sometimes been reported previously.
The Panel considered that the first issue to be determined was whether the matter was covered by the ABPI Code. If the research was conducted on behalf of a UK pharmaceutical company (whether directly or via a third party) then it would be covered by the ABPI Code. If a study was run by a non UK company but had UK involvement such as centres, investigators, patients etc it was likely that the Code would apply. The Panel appreciated the global nature of much pharmaceutical company sponsored clinical research and a company located in the UK might not be involved in research that came within the ABPI Code. It was a well established principle that UK pharmaceutical companies were responsible for the activities of overseas affiliates if such activities related to UK health professionals or were carried out in the UK.
Clause 21.3 of the Second 2012 Edition of the Code stated that companies must disclose details of clinical trials in accordance with the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature.
The relevant supplementary information stated that this clause required the provision of details about ongoing clinical trials (which must be registered within 21 days of initiation of patients enrolment) and completed trials for medicines licensed for use in at least one country. Further information was to be found in the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases 2009 and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature 2010, both at http://clinicaltrials.ifpma.org.
The Panel noted that the first Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases was agreed in 2005 by the International Federation of Pharmaceutical Manufacturers and Associations (IFPMA), the European Federation of Pharmaceutical Industries and Associations (EFPIA), the Japanese Pharmaceutical Manufacturers Association (JPMA) and the Pharmaceutical Research and Manufacturers of America (PhRMA). The announcement was dated 6 January 2005.
The Panel noted that Article 9, Clinical Research and Transparency, of the most recent update of the IFPMA Code of Practice (which came into operation on 1 September 2012) included a statement that companies disclose clinical trial information as set out in the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases (2009) and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature (2010). As companies had, in effect, agreed the joint positions their inclusion in the IFPMA Code should not have made a difference in practice to IFPMA member companies but meant that IFPMA member associations had to amend their codes to reflect Article 9. The Second 2012 Edition of the ABPI Code fully reflected the requirements of the IFPMA Code. The changes introduced in the ABPI Code were to update the date of the Joint Position on the Disclosure of Clinical Trial Information and to include the new requirement to disclose in accordance with the Joint Position on the Publication of Clinical Trial Results. Pharmaceutical companies that were members of national associations but not of IFPMA would have additional disclosure obligations once the national association amended its code to meet IFPMA requirements. The disclosures set out in the joint positions were not required by the EFPIA Codes.
The Panel noted that even if the UK Code did not apply many of the companies listed by the complainant were members of IFPMA and/or EFPIA.
The Panel considered that it was good practice for clinical trial results to be disclosed for medicines which were first approved and commercially available after 6 January 2005 (the date of the first joint position). This was not necessarily a requirement of the ABPI Codes from that date as set out below.
As far as the ABPI Code was concerned, the Panel noted that the first relevant mention of the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases 2005 was in the supplementary information to Clause 7.5 of the 2006 Code:
‘Clause 7.5 Data from Clinical Trials
Companies must provide substantiation following a request for it, as set out in Clause 7.5. In addition, when data from clinical trials is used companies must ensure that where necessary that data has been registered in accordance with the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases 2005.’
Clause 7.5 of the 2006 Code required that substantiation be provided at the request of health professionals or appropriate administrative staff. Substantiation of the validity of indications approved in the marketing authorization was not required. The Panel considered this was not relevant to the complaint being considered which was about disclosure of clinical trial results. The Joint Position 2005 was mentioned in the supplementary information to Clause 21.5 but this did not relate to any Code requirement to disclose clinical trial results.
In the 2008 ABPI Code (which superceded the 2006 Code and came into operation on 1 July 2008 with a transition period until 31 October 2008 for newly introduced requirements), Clause 21 referred to scientific services and Clause 21.3 stated:
‘Companies must disclose details of clinical trials.’
The relevant supplementary information stated:
‘Clause 21.3 Details of Clinical Trials
This clause requires the provision of details about ongoing clinical trials (which must be registered within 21 days of initiation of patients enrolment) and completed trials for medicines licensed for use in at least one country. Further information can be found in the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases 2005 (http://clinicaltrials.ifpma.org).
Details about clinical trials must be limited to factual and non-promotional information. Such information must not constitute promotion to health professionals, appropriate administrative staff or the public.’
In the 2011 Code (which superceded the 2008 Code and came into operation on 1 January 2011 with a transition period until 30 April 2011 for newly introduced requirements), the supplementary information to Clause 21.3 was updated to refer to the 2008 IFPMA Joint Position.
In the Second 2012 Edition (which came into operation on 1 July 2012 with a transition period until 31 October 2012 for newly introduced requirements), changes were made to update the references to the joint position and to include the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature.
Clause 21.3 now stated:
‘Companies must disclose details of clinical trials in accordance with the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature.’
The relevant supplementary information stated:
‘Clause 21.3 Details of Clinical Trials
This clause requires the provision of details about ongoing clinical trials (which must be registered within 21 days of initiation of patients enrolment) and completed trials for medicines licensed for use in at least one country. Further information can be found in the Joint Position on the Disclosure of Clinical Trial Information via Clinical Trial Registries and Databases 2009 and the Joint Position on the Publication of Clinical Trial Results in the Scientific Literature 2010, both at http://clinicaltrials.ifpma.org.
Details about clinical trials must be limited to factual and non-promotional information. Such information must not constitute promotion to health professionals, appropriate administrative staff or the public.’
The Panel noted that in the 2014 ABPI Code the disclosure requirements which had previously been stated in Clause 21 had been moved to Clause 13. In addition, the supplementary information stated that companies must include on their website information as to where details of their clinical trials could be found. The 2014 Code would come into effect on 1 May 2014 for newly introduced requirements following a transition period from 1 January 2014 until 30 April 2014.
The Panel examined the Joint Position on the Disclosure of Clinical Trial Information which was updated on 10 November 2009 and superseded the Joint Position 2008. With regard to clinical trial registries the document stated that all trials involving human subjects for Phase I and beyond at a minimum should be listed. The details should be posted no later than 21 days after the initiation of enrolment. The details should be posted on a free publicly accessible internet-based registry. Examples were given. Each trial should be given a unique identifier to assist in tracking. The Joint Position 2009 provided a list of information that should be provided and referred to the minimum Trial Registration Data Set published by the World Health Organisation (WHO). The Joint Position 2009 referred to possible competitive sensitivity in relation to certain data elements and that, in exceptional circumstances, this could delay disclosure at the latest until after the medicinal product was first approved in any country for the indication being studied. Examples were given.
The Panel noted that the complaint related to the disclosure of clinical trial results.
With regard to the disclosure of clinical trial results the Joint Position 2009 stated that the results for a medicine that had been approved for marketing and was commercially available in at least one country should be publicly disclosed. The results should be posted no later than one year after the medicine was first approved and commercially available. The results for trials completed after approval should be posted one year after trial completion – an adjustment to this schedule was possible to comply with national laws or regulations or to avoid compromising publication in a peer-reviewed medical journal.
The Joint Position 2009 included a section on implementation dates and the need for companies to establish a verification process.
The Joint Position 2005 stated that the results should be disclosed of all clinical trials other than exploratory trials conducted on a medicine that was approved for marketing and was commercially available in at least one country. The results generally should be posted within one year after the medicine was first approved and commercially available unless such posting would compromise publication in a peer-reviewed medical journal or contravene national laws or regulations. The Joint Position 2008 was dated 18 November 2008 and stated that it superseded the Joint Position 2005 (6 January and 5 September). The Joint Position 2008 stated that results should be posted no later than one year after the product was first approved and commercially available in any country. For trials completed after initial approval these results should be posted no later than one year after trial completion. These schedules would be subject to adjustment to comply with national laws or regulations or to avoid compromising publication in a peer reviewed medical journal.
The Joint Position on the Publication of Clinical Trial Results in the Scientific Literature was announced on 10 June 2010. It stated that all industry sponsored clinical trials should be considered for publication and at a minimum results from all Phase III clinical trials and any clinical trials results of significant medical importance should be submitted for publication. The results of completed trials should be submitted for publication wherever possible within 12 months and no later than 18 months of the completion of clinical trials for already marketed medicines and in the case of investigational medicines the regulatory approval of the new medicine or the decision to discontinue development.
Having examined the various codes and joint positions, the Panel noted that the Joint Position 2005 excluded any clinical trials completed before 6 January 2005. The position changed on 18 November 2008 as the Joint Position 2008 did not have any exclusion relating solely to the date the trial completed. The Joint Position 2009 was similar to the Joint Position 2008 in this regard.
The Panel noted that deciding which Code applied, and thus which joint position, was complicated. It noted that the 2011 Code which, taking account the transition period, came into operation on 1 May 2011 was the first edition of the Code to refer to the Joint Position 2008.
The Panel concluded that from 1 November 2008, (allowing for the transition period) until 30 April 2011 under the 2008 Code companies were required to follow the Joint Position 2005. From 1 May 2011 until 31 October 2012 under the 2012 Code companies were required to follow the Joint Position 2008. Since 1 November 2012 companies were required to follow the Joint Position 2009. The Panel considered that since the 2008 Code companies were, in effect, required to comply with the Joint Position cited in the relevant supplementary information. The relevant supplementary information gave details of what was meant by Clause 21.3 (Clause 13.1 in the 2014 Code). The Panel accepted that the position was clearer in the Second 2012 Edition of the Code. The Panel noted that the 2011 Code should have been updated to refer to the Joint Position 2009.
For medicines first licensed and commercially available in any country from 1 November 2008 until 30 April 2011 the results of clinical trials completed before 6 January 2005 would not have to be posted.
From 1 May 2011 there was no exclusion of trials based solely on completion date and so for a product first licensed and commercially available anywhere in the world after 1 May 2011 the applicable joint positions required relevant clinical trial results to be posted within a year of the product being first approved and commercially available or within a year of trial completion for trials completed after the medicine was first available.
Noting that the complaint concerned licensed products the Panel considered that the trigger for disclosure was the date the product was first approved and commercially available anywhere in the world. This would determine which version of the Code (and joint position) applied for trials completed prior to first approval. The next consideration was whether the trial completed before or after this date. For trials completing after the date of first approval, the completion date of the trial would determine which Code applied. The Panel considered that the joint positions encouraged disclosure as soon as possible and by no later than 1 year after first availability or trial completion as explained above. The Panel thus considered that its approach was a fair one. In this regard, it noted that the complaint was about whether or not trial results had been disclosed, all the joint positions referred to disclosure within a one year timeframe and companies needed time to prepare for disclosure of results. The Panel considered that the position concerning unlicensed indications or presentations of otherwise licensed medicines etc would have to be considered on a case by case basis bearing in mind the requirements of the relevant joint position and the legitimate need for companies to protect intellectual property rights. The Panel followed the decision tree set out below which it considered set out all the relevant possibilities.
During its development of the decision tree, the Panel sought advice from Paul Woods, BPharm MA (Medical Ethics and Law) of Paul Woods Compliance Ltd who provided an opinion. Mr Woods was not provided with details of the complaint or any of the responses. The advice sought was only in relation to the codes and joint positions.
The Panel considered the complaint could be read in two ways: firstly that the companies listed had not disclosed the data referred to in the CMRO publication relating to the products named or secondly, more broadly, that the companies had not disclosed the clinical trial data for the product named ie there could be studies in addition to those looked at in the CMRO publication. The Panel decided that it would consider these cases in relation to the studies covered by the CMRO publication and not on the broader interpretation. Companies would be well advised to ensure that all the clinical trial results were disclosed as required by the Codes and joint positions. The Panel considered that there was no complaint about whether the results disclosed met the requirements of the joint positions so this was not considered. In the Panel’s view the complaint was only about whether or not study results had been disclosed and the timeframe for such disclosure.
The CMRO publication stated that as far as the IFPMA Joint Position was concerned implementation had been somewhat variable in terms of completeness and timing. The Panel noted that a number of studies were referred to in the CMRO publication as ‘unevaluable’ and these were not specifically mentioned by the complainant. The CMRO publication focussed on the disclosure of evaluable trial results and the Panel only considered those evaluable trials.
The Panel noted that its consideration of these cases relied upon the information provided by the respondent companies. The CMRO publication did not identify the studies evaluated; it only provided quantitative data. The Panel noted that the study ran from 27 December 2012 to 31 January 2013 and was published in November 2013. The Panel considered that companies that might not have been in line with various disclosure requirements had had a significant period of time after the study completed and prior to the current complaint being received to have disclosed any missing information. It appeared that the authors of the CMRO publication had contacted various companies for additional information.
The Panel noted that the case preparation manager raised Clause 1.8 of the Second 2012 Edition with the companies. The supplementary information to Clause 1.8, Applicability of Codes, inter alia, referred to the situation when activities involved more than one country or where a pharmaceutical company based in one country was involved in activities in another country. The complainant had not cited Clause 1.8. The Panel noted that any company in breach of any applicable codes, laws or regulations would defacto also be in breach of Clause 1.8 of the Code; the converse was true. The Panel thus decided that as far as this complaint was concerned, any consideration of a breach or otherwise of Clause 1.8 was covered by other rulings and it decided, therefore, not to make any ruling regarding this clause (or its equivalent in earlier versions of the Code).
PANEL RULING IN CASE AUTH/2657/11/13
The Panel noted the CMRO publication in that twenty-nine Iressa studies had not been disclosed in the timeframe. The disclosure percentage was 56%. Twelve studies had not been disclosed giving a disclosure percentage at 31 January 2013 for trials completed at 31 January 2012 of 84%. A footnote stated that the majority of Phase II/III trials were completed prior to FDAAA 801 requirements. The remaining undisclosed trials were in the process of publication.
The Panel noted AstraZeneca’s submission regarding the studies. It noted that AstraZeneca was a UK registered company. It could be argued that this meant the UK Code applied as the studies were in effect run by a UK company.
Decision Tree – for expanded version see Case Report PDF
Developed by the Panel when considering the complaint about the disclosure of clinical trial results
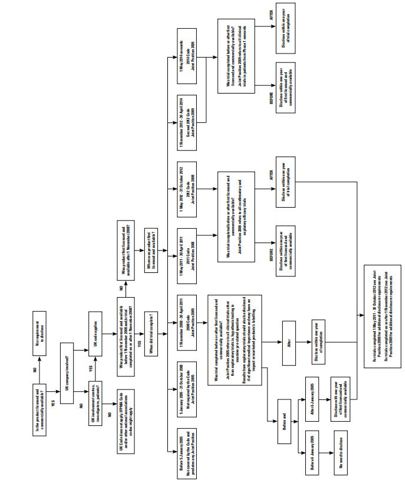
The Panel agreed with AstraZeneca that it was not responsible for disclosure of investigator-sponsored studies (15 trials). It was good practice for a company to strongly advocate publication of such data but the Code and joint positions only related to pharmaceutical company sponsored studies. Thus the Panel ruled no breach of the Code as the matter was not within the scope of the Code.
The Panel noted that AstraZeneca first got a marketing authorization for Iressa in Japan in 2002 and in the Panel’s view, this was when the company first became responsible for meeting any disclosure requirements. The first joint position (January 2005) was not referred to in the Code until the 2008 Code which was effective from 1 November 2008. Thus any Iressa trials completed before this data were not required to be disclosed under the Code.
The Panel noted that of the remaining 38 trials (53 minus 15 investigator-sponsored trials), 35 were Phase I, exploratory Phase II or Phase III studies all of which completed before 1 November 2008. In that regard, there was no requirement under the Code to disclose these studies. The Panel thus ruled no breach of Clause 21.3 of the 2008 Code and consequently no breach of Clauses 9.1 and 2. (The Panel noted that although there was no requirement under the Code to do so, the results for 23 of these trials had been disclosed).
An AstraZeneca Thailand non-interventional study completed in August 2010, which was after Iressa was first approved and commercially available. The Panel noted AstraZeneca’s submission that these results were disclosed on its own website in November 2010. The study was a retrospective cohort study on patients from two tertiary hospitals in Thailand. It was not an interventional study, it was not clear whether there was any UK involvement and the Joint Position 2005 appeared not to require disclosure of the results of a non interventional trial. (In the Joint Position 2009 it was clear that only the results from interventional studies had to be disclosed). In any event the results had been disclosed publicly within one year and thus the Panel ruled no breach of Clauses 2, 9.1 and 21.3 of the 2008 Code.
The Panel noted that the results from two trials remained undisclosed – an AstraZeneca Canada study which completed in August 2011 and an AstraZeneca Taiwan study which completed in August 2009. AstraZeneca submitted that the publication of the results was expected.
The Panel considered that although AstraZeneca was a UK registered company, the company’s arrangements were such that it was clear that the responsibility for disclosure was with the local company. It considered that the matter was potentially covered by the UK Code but as the responsibilities had been made very clear in a company SOP it ruled no breach of Clause 21.3 of the 2008 Code in relation to the AstraZeneca Taiwan trial and no breach of Clause 21.3 of the 2011 Code in relation to the AstraZeneca Canada study. The Panel consequently ruled no breaches of Clauses 9.1 and 2 of the respective Codes.
Complaint received 21 November 2013
Case completed 20 March 2014
see cases: 3005,2908,2906,2903,2898,2763,2676,2674,2673,2672,2671,2670,2669,2667,2666,2665,2664,
2663,2662,2661,2659,2657,2654